Using deep learning diffusion models for denoising of low-coverage RNA-seq data
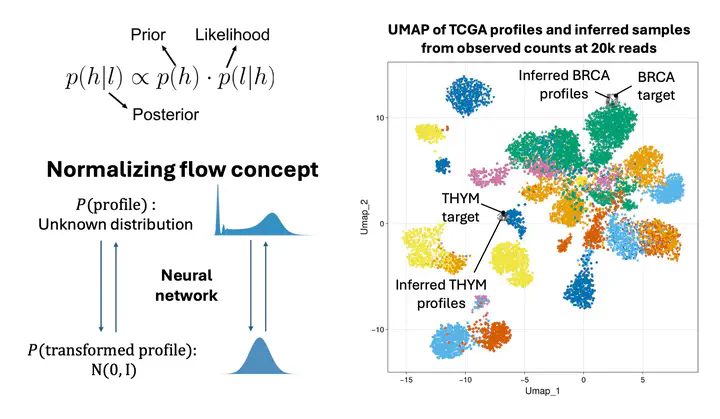
Although RNA sequencing (RNA-seq) allows us to gain deeper insight into human biology, the number of samples that can be sequenced is still limited by sequencing costs. The cost per sample can be reduced by decreasing the sequencing depth, but this leads to lower-quality data. Additionally, existing techniques to artificially increase data quality, such as imputation methods in single-cell RNA-seq, are not suitable for denoising standard RNA-seq data (bulk RNA-seq).
One-to-one denoising neural networks were implemented and allowed for the recovery of most information lost from low-depth RNA-seq. Unfortunately, these do not encode the uncertainty and noise related to decreased sequencing depth. As such, we are exploring probabilistic inference models, such as Bayesian inference and variational inference, while leveraging Normalizing Flows and copulas, to instead predict the distribution of true transcriptomic profiles that could have originated the observed low-depth counts.
This new paradigm has the potential to create new sequencing standards for RNA-seq in various experimental, analytical or hospital areas, making it possible to both reduce costs and provide large quantities of data.